Anorectal malformations are the collective term for a group of conditions in which the terminal portion of the digestive tract, namely the rectum and the anus, do not develop properly while the baby is in the womb. As a result, these children are born without a normal anus.
The job of the rectum and the anus is to store waste material (stool) and expel it from the body. In the absence of a properly formed anus and rectum, the waste material keeps collecting in the intestines leading to complications. These complications can be completely avoided by prompt surgical interventions.
What are the types of anorectal malformations?
Anorectal malformations appear differently in boys and girls.
In boys, the terminal part of the digestive tract may open into the urethra near the prostate forming a recto-prostatic fistula, or into the bulbar urethra forming a recto-bulbar fistula. These fistulae are very narrow and cannot expel stool. Sometimes these boys may expel a tiny drop of meconium (the baby’s very first stool) at the urethra, or they may pass urine mixed with meconium (meconuria).
Sometimes the malformation can occur at a higher level, and the digestive tract can open into the urinary bladder at its neck. This is called a bladder neck fistula. These malformations can be challenging to treat as the bladder neck plays an important role in urinary continence.
Still another type of anorectal malformation results in the digestive tract ending in a blind pouch with no communication to any other part. Alternatively, the digestive tract can open on the perineum through a small, abnormal aperture called a perineal fistula. Finally, the anus and the rectum may develop to reach the usual anal site, but they may be too narrow (anorectal stenosis) or blocked somewhere along their length (rectal atresia).
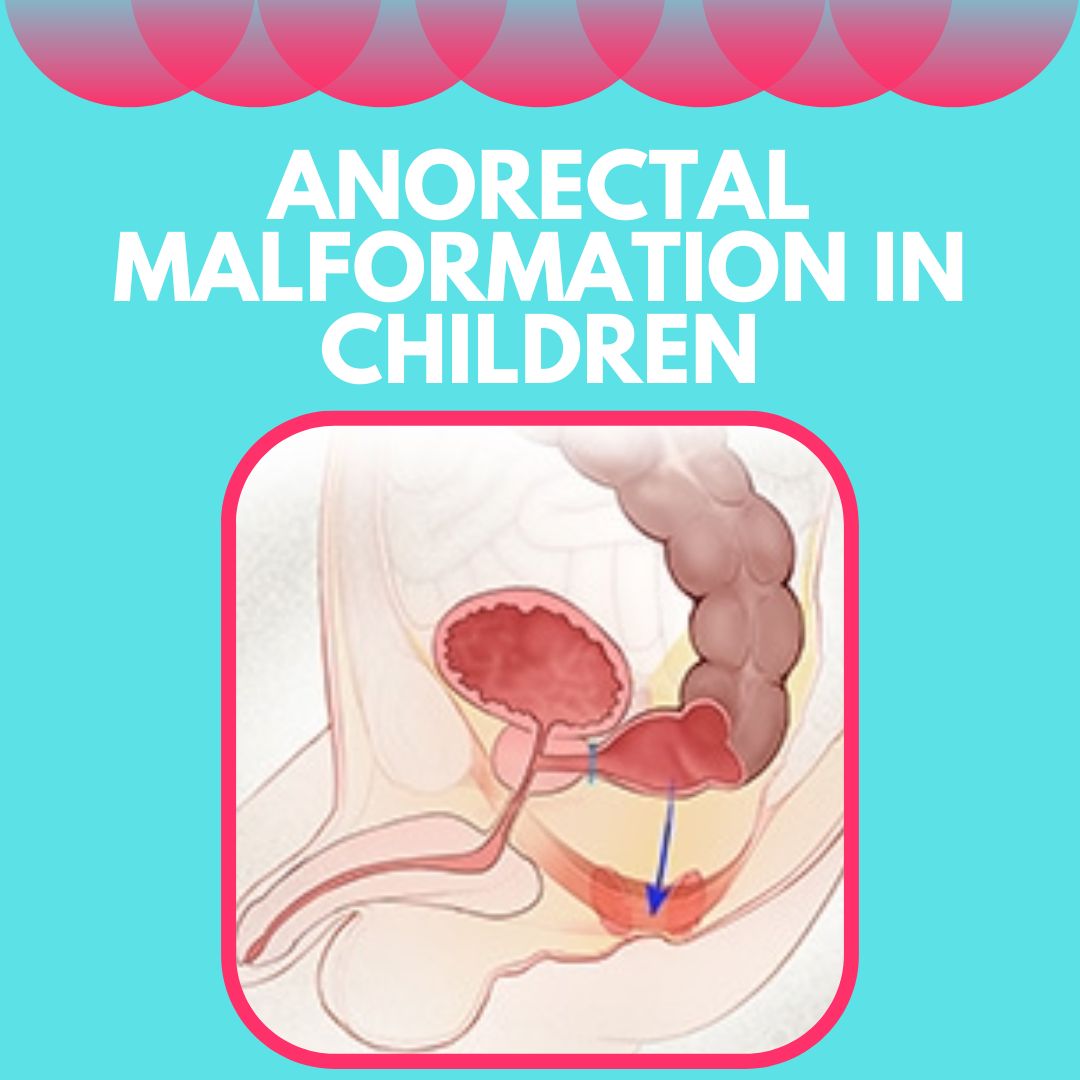
In girls, the most common anorectal malformation is a vestibular fistula. In this type of malformation, the digestive tract ends in a tiny, abnormal fistula that opens just behind the vagina, sharing a wall with it. Perineal fistulas, blind-ending malformations with no fistula, anorectal stenosis, and rectal atresia may also occur in girls as they do in boys.
The anatomy of a vestibular fistula type of anorectal malformation in girls. The rectum opens through a fistula just behind the vagina instead of passing through the sphincter muscle complex to form a normal anus.
A complex anomaly that occurs in girls is the cloaca. In this malformation, the digestive tract, the urinary tract, and the reproductive tract open into a single common channel. Such children have a single opening in their perineum for meconium, urine, and mucus from the uterine tract to exit. Cloaca is very commonly associated with malformations of the lower urinary tract, vagina, and uterus.
Identifying the type of anorectal malformation is important to plan surgical repair. The pediatric surgeon orders a series of tests to determine the type of malformation. In many cases, the exact anatomy of the malformation is determined just before or as a part of reconstructive surgery.
Can there be other conditions in the baby along with Anorectal malformations (ARM)?
At least 50% of children born with an ARM have some associated congenital condition. The most common systems to be affected in these children are the urinary system (kidneys, ureters, and bladder), the heart, the spine, and the food pipe. A number of children also have limb deformities.
All babies found to have an ARM are screened for these conditions and treated accordingly. Some of the common conditions are:
- Vesicoureteral reflux (urinary system)
- Ventricular Septal Defect (heart)
- Hemivertebrae (spine)
- Tracheoesophageal fistula and esophageal atresia (food pipe).
This association of anomalies is often called VACTERL association or VACTERL Syndrome, wherein VACTERL stands for Vertebral, Anorectal, Cardiac, Tracheo-Esophageal, Renal, and Limb. A child is said to have the syndrome if at least three of these defects are present together.
What happens if my newborn baby has anorectal malformations?
Most of the time, the pediatrician attending the delivery will examine the baby to rule out any anorectal malformation. If your baby is found to have an anorectal malformation, you will be instructed not to feed the baby until the pediatric surgeon has seen the child. Rarely, the condition may come to the attention of the parents first. In either case, a pediatric surgeon needs to be called in at the earliest.
The pediatric surgeon will examine the baby physically and order X-rays. He or she will also look for associated anomalies, especially esophageal atresia as it needs immediate attention. The immediate priority is to create an outlet for the pent-up intestinal contents, failing which the intestines could rupture leading to a life-threatening situation. Findings from the physical exam and the X-rays will help the surgeon decide on one of the following:
a) Immediate corrective surgery- This is possible in certain types of malformations where the digestive tract has developed almost all the way to the anus. The surgeon will perform the surgery when the baby is at least 24 hours old. An anus is created at its normal position.
b) Delayed corrective surgery- in malformations such as vestibular fistulae or perineal fistulae, the opening may be big enough to allow the baby’s meconium and first milk stool to pass easily, but too small for formed stool. In such a case, the operation can be deferred till the baby gains some weight. Daily gentle dilation of the fistula with a feeding tube may be required till the operation.
c) Colostomy construction followed by delayed corrective surgery- this plan of action is followed in boys with recto prostatic or retrobulbar fistula or blind pouches and in girls with cloaca or blind pouches. Boys and girls with rectal atresia also need this line of treatment. First, the surgeon creates a colostomy which is an opening of the large intestine onto the abdominal wall. The baby’s meconium and stool are expelled through this opening and collected in a colostomy bag. This temporary outlet for intestinal waste and stool is created so that the baby can feed and grow. When the baby has gained sufficient weight, corrective surgery is performed. A third surgery is then needed to close the colostomy.
What are the various corrective surgeries that can be performed?
The type of corrective surgery ultimately performed depends on the anatomy of the malformation. The goal of all these surgeries is to create a new anus that lies at the center of the sphincter muscle complex, which is a group of muscles that help us control our bowel movements. Placement of the new anus and rectum at the center of this muscle complex is critical for the future continence of the child. These surgeries include:
PSARP– The Posterior Sagittal AnoRectoPlasty or PSARP, first described by Dr. Alberto Peña (pronounce peh-nyah) in 1982, has revolutionized surgery for anorectal malformations. For this surgery, the patient is positioned face-down and frog-legged on the operating table. The buttock muscles are split in the midline and the bowel is mobilized and brought to the center of these muscles. The bowel is then fixed in place and the muscles are closed around it. There is no abdominal incision for this surgery. It can be performed for blind pouches without a fistula, recto-prostatic fistulae, recto-bulbar fistulae, perineal fistulae, and vestibular fistulae.
ASARP– Described by Okada and Kamata in 1992, the Anterior Sagittal AnoRectoPlasty is similar in principle to the PSARP but is performed with the child lying flat on the back and frog-legged. The buttock muscles are split in the midline but this splitting is limited as compared to PSARP. It can be performed for vestibular fistulae and perineal fistulae. In these types of malformations, the decision on whether an ASARP or a PSARP is used depends on the surgeon’s experience with the two methods. The procedures are anatomically essentially the same, as are their outcomes.
Laparoscopic Abdominoperineal Pull Through– This surgery is performed for high fistulae such as bladder neck fistulae. The fistula is tied off through the abdomen and the bowel is mobilized laparoscopically. The buttock muscles are then split in the midline and the bowel is placed in the center. This procedure can be performed by conventional surgery as well giving an incision on the abdomen.
PSAVURP– The Posterior Sagittal AnoRectoVaginoPlasty is a complex operation performed to correct the cloaca. It is similar to PSARP in its approach. Depending on the anatomy of the malformation, supplemental procedures may be required to reconstruct the genito-urinary tract. Thus the reconstruction may either be staged or performed in one go.
How do I care for my baby after surgery?
If your baby has been given a colostomy, you will be taught how to care for the colostomy at home. The colostomy will not be closed until after the corrective surgery.
After corrective surgery, if your child has a colostomy, you may be allowed to resume feeding a few hours after surgery. In the absence of an ostomy, feeding may have to be withheld for a few days. In such a case, your child will be given the required nutrition through an intravenous drip. You will be taught how to care for the wound and how to keep it clean.
About two weeks after surgery, you will be taught to dilate the new anus. This procedure is important to make sure that the anus does not tighten up and close off. A dilator that fits snugly into the anus is passed twice a day. The size of the dilator is increased till the anus has become an appropriate size. If your child has a colostomy, this is the time when it can be closed. The dilation has to be continued after the closure of the colostomy for at least a year, although the frequency of dilation is reduced. The size of the dilator to use and the frequency of dilation will be monitored by the surgeon.
Follow-up with the surgeon is necessary at least till your child has toilet trained. Annual follow-up till your child is an adult is desirable. Monitoring/ treatment of associated conditions, if any, will continue as well. Once your baby has been weaned, he or she may have to follow some dietary modifications to avoid constipation.
Will my baby toilet train normally after the surgery?
Children with anorectal malformation find it more difficult to toilet train than others for a variety of reasons. Be prepared for your child to achieve continence at a later age than his or her peers. These children are also very prone to constipation, so be careful not to mistake constipation for early continence. The potential for continence in the child depends mainly on the anatomy of the malformation. The surgeon will be able to explain this to you during your early visits.
However, if your child is unable to wear normal underwear by the age of five years, there are a number of surgical and nonsurgical methods to help him or her achieve a level of continence that allows him or her to enjoy normal social interactions. We have a bowel management program that focuses on a combination of diet and washouts to help keep the child clean. Surgical procedures such as a MACE (Malone’s antegrade colonic enema) can be performed to help the child take care of themselves. Most children with anorectal malformations do achieve a level of continence that allows them to lead a full, productive life.
What is the long-term outlook for my child?
Most children with anorectal malformation grow up to live independent productive lives. The presence and severity of associated conditions are major determining factors though. In the absence of a severe associated condition, these children develop greater insight into their own continence mechanisms and are able to participate more fully in their bowel management. They are usually able to have normal social and professional interactions. Girls born with an anorectal malformation may have associated structural anomalies in their uterus which might become significant at the time of menarche. These anomalies are all treatable. Some women born with anorectal malformation may need assisted reproductive techniques at the time of starting a family, and childbirth is usually by cesarean section, given that their perineal body has undergone reconstruction. All in all, anorectal malformations have a good prognosis today, and the future is even more hopeful.
About Dr. Geeta Kekre
Dr. Geeta Kekre is a Paediatric Urologist in Pune She is a specialist in pediatric minimal access surgery including robotic surgery as well as reconstructive pediatric urology. After ten years in Mumbai, Dr. Kekre returned to her hometown bringing with her a passion for the surgical care of children and a distinct set of skills in the areas of pediatric minimal access surgery, pediatric endourology, fetal hydronephrosis and antenatal parental counseling, pediatric incontinence, and voiding dysfunction. Dr. Geeta Kekre has a keen interest in clinical academics with over 42 publications in various international journals, including 9 original articles and 32 rare case reports. Her focus is on tailoring surgical therapy to allow her patients to have as active and fun a childhood as possible while delivering the best achievable medical outcomes.